主办单位:中国民族卫生协会基层卫生人才工作委员会 All Rights Reserved
技术支持单位:北京天元中和医药科技研究院
京ICP备09057878号-6
理论研究
当前位置:首页 > 理论研究 恶性萎缩性丘疹病一例报告
作者:河南京城皮肤中医院 阅读: 54 次 来源:基层卫生人才工作委员会
李浩 刘高岗 王豫平 狄梅 田新科
河南京城皮肤中医院
临床资料:患者,男,67岁
主诉:全身反复起水肿性丘疹、溃疡、坏死4年余。
现病史:4年前无明显诱因,患者躯干部起圆形,光滑,坚实的半球形丘疹,很快变平,坏死中心凹陷呈脐窝状,形成不规则斑片,损害中央有溃疡坏死,局部有黄色脓液,愈合后发生皮肤萎缩,周边留下线状边缘,患处无自觉症状,2月后泛发全身,曾在外院诊治,检查血常规、肝功能、肾功能、凝血功能、梅毒、艾滋病未见异常,在院给予双嘧达莫50mg,3次/日,阿司匹林肠溶片25mg,3次/日,外用复方多粘菌素B软膏等药物,症状稍缓解,为求进一步治疗来我院。
既往史、个人史及家族史:患者否认高血压、高血脂、糖尿病及冠心病等其他慢性疾病史,否认肝炎、结核等传染病及密切接触史,否认药物和食物过敏史,否认皮损处局部外伤史,否认非婚性接触史。家族中无类似疾病患者。
体格检查:各系统检查均正常。
皮肤科检查:全身见红色水肿性丘疹,呈光滑坚实的半球形,局部出现坏死性中心凹陷,呈脐窝状,皮损处有萎缩。
实验室检查:血常规、尿常规、便常规、肝肾功能、免补七项、传染病四项、血糖、血脂七项、ASO、ESR、电解质、血凝四项等未见异常。
切除皮损组织行病理检查:取右上肢皮损,示表皮萎缩,真皮胶原纤维肿胀,微动脉血管内膜细胞肿胀,部分管壁破坏,偶见血栓形成,未见皮下组织。
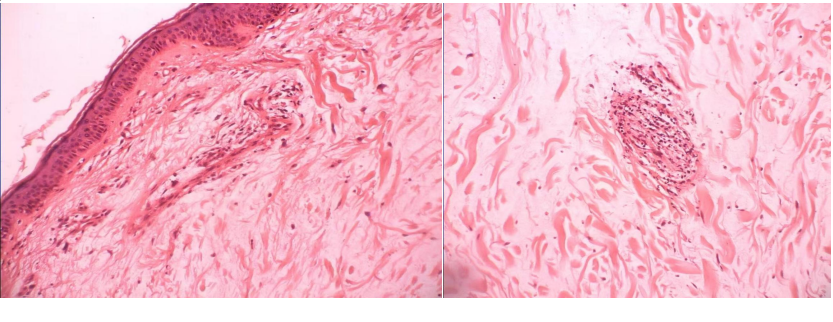
诊断:恶性萎缩性丘疹病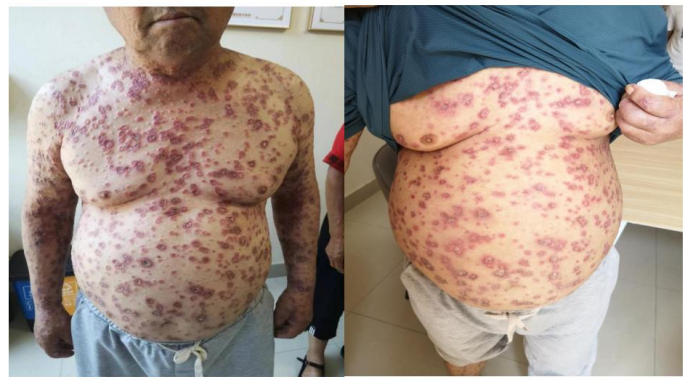
↑治疗前 ↑治疗后
治疗:门诊给予雷公藤多苷片20mg,3次/日口服,复方甘草酸苷片50mg ,3次/日,外用牛碱性成纤维细胞生长因子凝胶、氧化锌软膏,治疗一个月后症状明显减轻,溃疡处基本愈合,躯干部位皮损红消退。
讨论: 恶性萎缩性丘疹病由 Kohlmeier 于 1941 年首次报道,1942 年由 Degoes 正式命名为 MAP,是 一种罕见的累及中、小型动脉的血管闭塞性疾病[1]患处皮损早期为淡红色斑,颜色在很短的时间内变成红色圆形,光滑坚实的半球形丘疹,丘疹很快变平,坏死,中心凹陷呈脐窝状,形成不规则斑,坏死,中央有溃疡。皮损主要累及皮肤、胃肠道和中枢神经系统,后期多损害胃肠道,出现缺血性梗塞发生于肠道可有急腹症的症状,包括剧烈腹痛、腹胀、消化不良、发热、呕血、腹膜炎、肠穿孔多发性穿孔所导致的爆发性腹膜炎可引起死亡。是本病常见的死亡原因,此例患者经过8个月随访,死于多发性肠道穿孔。平时住院查房时已宣教,提示患者本人及家属此病有多发性肠穿孔的风险。患者来我院之前已发生过肠道小面积穿孔。在开封市人民医院手术治疗,得到控制。恶性萎缩性丘疹病是由小中动脉闭塞引起的进行性血管病和血管内皮炎,是一种致死性皮肤肠道闭塞性动脉炎综合征,本病主要累及皮肤胃肠道,偶也可常累及神经系统,在临床上本病发病率很低,病因不明,可能与凝血异常,纤维溶解抑制功能异常,自身免疫,及病毒感染有关,也有报道与遗传有关,在治疗上还没有治疗恶性丘疹病的统一治疗方法,偶有报道阿司匹林单独或配合已酮可可碱应用对患者有效[2]。笔者通过服用复方甘草酸苷联合雷公藤多苷片,加外用药物治疗,皮损症状缓解,雷公藤多苷片有较强的抗炎及免疫抑制作用,在抗炎作用方面,它能拮抗和抑制炎症介质的释放,同时能抑制体液免疫和细胞免疫,复方甘草酸苷和雷公藤多苷片有协同作用,共同抗炎调节免疫,患者皮损明显减轻,预后本病不经治疗平均存活2年左右,当病变累及消化道出现急腹症时手术治疗是首选,目前在治疗恶性萎缩性丘疹病时,药物治疗没有特效性的治疗药物,临床常用药物如下:1.纤溶和免疫抑制治疗 药物选择包括雷公藤多苷片、环孢素 A、硫唑嘌呤、环磷酰胺和皮质类固醇等,只能改善皮损症状,对内脏病变无效。GUO等[3]报道了1例小儿神经性恶性萎缩性丘疹病对皮质类固醇联合静脉注射免疫球蛋白 (IVIG)的治疗方案反应良好。2.抗凝治疗和抗血小板治疗 药物选择包括 乙酰水杨酸、双嘧达莫、乙酮可可碱、替氯地平、肝素 等,可能可以在血栓形成前阻止血小板聚集,抑制疾 病进展,因此被选作新诊断的恶性萎缩性丘疹病患者的首选治疗方法,但这些药物对于其他有全身系统性症状的 治疗效果仍不明确[4]。3.随着对发病机制的深入研究,也出现了一些新药,如依库珠单抗、MAGRO等[5],使用依库珠单抗治疗1例患者,发现该患者治疗后无活动性内出血,依库珠单抗和曲前列腺素联合治疗。4.利妥昔单抗和环磷酰胺联合,Day 等[6]等通过利妥昔单抗和环磷酰胺联合治疗成功治疗了一例类似晚期 degos 病并伴有胃肠穿孔的青少年皮肌炎。这种疑难病例是我们临床工作需要认真学习,及时做好患者后期的回访工作,加强对目前治疗方案和用药的探索。提高患者的生活质量,为自己以后的临床工作增加更多的经验。特别是这种罕见病,加强收集资料,整理完整。
[1]Theodorids A,Makrantonaki E,Zouboulis CC. Malignant atrophic papulosis( Kohlmeier-Degos disease) -a review[J]. Orphanet J Rare Dis,2013,8( 1) : 10.
[2]Vicktor C,Schultz-Ehrenburg U.Malignant atrophic papulosis (Kohlmeier-Degos): diagnosis, therapy and course [German]. Hautarzt.2001;52:734-7.
[3]GUO YF,PAN WH,CHENG RH,et al. Successful treatment of neurological malignant atrophic papulosis in child by corticoste⁃ roid combined with intravenous immunoglobulin[J]. CNS Neuro⁃ sci Ther,2014,20(1):88-91.
[4] GUPTA S,DOGRA S,SAIKIA UN,et al. Degos disease with dermatomyositis- like phenomenon:a diagnostic dilemma and a therapeutic challenge[J]. J Cutan Med Surg,2011,15(3):
162-166.
[5]MAGRO CM,WANG X,GARRETT-BAKELMAN F,et al. The effects of Eculizumab on the pathology of malignant atrophic papu⁃ losis[J]. Orphanet J Rare Dis,2013,8:185.
[6]DAY W,GABRIEL C,KELLY RE JR,et al. Juvenile dermato⁃ myositis resembling late-stage Degos disease with gastrointestinal perforations successfully treated with combination of cyclophos⁃ phamide and rituximab:case-based review[J]. Rheumatol Int, 2020,40(11):1883-1890.